What Was Published
A team led by Hiroaki Suga at the University of Tokyo, working with Junichi Takagi's group at the Institute for Protein Research at Osaka University, has published a method that changes how peptide drug candidates can be selected against complex membrane receptors. The paper appeared in Angewandte Chemie International Edition on March 12, 2026, with the formal title "Direct Selection of Functional De Novo Macrocycles for Activation of On-Cellulo Insulin Receptor." It carries the DOI 10.1002/anie.202526008, sits on PubMed at PMID 41815006, and was featured by the American Peptide Society on May 12, 2026 as "Function-First Macrocycles".
The headline result is a single macrocyclic peptide named HL4 that binds and activates the human insulin receptor on intact cells, and a homodimer construct built from it with a serum half-life of around 51 hours. The deeper result is the platform itself: a variant of Suga's RaPID mRNA-display system that screens trillions of cyclic peptides against receptors in their native cellular environment, rather than against purified domains stuck to a plate. The platform is called RaPID-ExCells.
Key Facts
- Paper: Kuo Y-H, Mihara E, Takagi J, Suga H. Angew. Chem. Int. Ed. 2026 Mar 12. DOI 10.1002/anie.202526008.
- Platform: RaPID-ExCells — mRNA display of cyclic peptide libraries against intact mammalian cells, with detergent lysis preserving receptor conformation and co-immunoprecipitation for recovery.
- Target in this study: Human insulin receptor (IR), full-length, on HEK293 cells.
- Hit: HL4, a cyclic peptide. Kd around 132 nM. EC50 for IR autophosphorylation around 720 nM as a monomer.
- Function-first finding: Of 11 top affinity-selected peptides, only HL4 activated the receptor. Several non-functional peptides bound more tightly than HL4.
- Pharmacophore: An FYLWF aromatic motif at positions 9 to 13 is essential for IR activation. The motif mirrors the FYXWF sequence in earlier linear insulin-mimetic peptides such as Novo Nordisk's S597.
- Engineering: Deep mutational scanning using 66 amino acids, including 47 non-proteinogenic residues. para-methyl- and para-fluoro-phenylalanine at position 9 raised activity roughly 1.5-fold. D-tyrosine at position 5 was tolerated.
- Homodimer: Two HL4 monomers linked by PEG11 spacers to an L-Dap core. EC50 for IR autophosphorylation around 460 nM. Roughly 2.5-fold autophosphorylation at 1 µM compared with the monomer. Serum half-life around 51 hours.
- Selectivity: IR over IGF-1R despite the close structural relationship between the two receptors.
- Generality of the platform: RaPID-ExCells in principle works against any cell-surface receptor or membrane protein that can be recovered by co-immunoprecipitation.
Why This Paper Is a Big Deal Even If You Have Never Heard of It
Most drug screens for peptides that hit cell-surface receptors are run against the receptor's extracellular domain. The domain is cloned, expressed in E. coli or insect cells, purified, and pinned to a surface. A library of peptides washes over the surface. The peptides that stick get sequenced. Then a chemist makes the top hits and tests them on real cells to see if any of them do anything.
This works. It also fails in a particular way. Many receptors only fold correctly, or only present the binding surface a drug needs to hit, when they are sitting in a cell membrane with their full transmembrane and cytoplasmic regions attached and the right lipid environment around them. The insulin receptor is one of the harder cases. It is a constitutive dimer with two extracellular alpha subunits and two transmembrane beta subunits joined by disulfide bonds. Its activated conformation involves rearrangement of the dimer, not just occupancy of a site. A peptide that sticks tightly to a chunk of purified IR ectodomain may or may not engage the receptor in a way that produces signaling once you put it back on a cell.
RaPID-ExCells skips the purification step. The library — on the order of 10^12 to 10^13 distinct cyclic peptides — is panned directly against HEK293 cells overexpressing the full-length receptor. The cells are then gently lysed with a mild nonionic detergent that keeps the receptor's quaternary structure intact. The receptor, with whatever peptides it has captured, is pulled down with an antibody. The bound peptides go to next-generation sequencing. Suga's group has been moving the RaPID system in this direction for years; this paper is the cleanest demonstration on a receptor that defeats easier screens.
The Function-First Insight
Here is the part that should be on the wall of every drug-discovery lab working with peptides. The Tokyo and Osaka teams selected 11 of their top affinity-enriched cyclic peptides from the screen. They made them. They measured binding to the insulin receptor by surface plasmon resonance. They measured receptor activation by autophosphorylation on cells.
Ten of the 11 peptides bound the receptor. Some bound very tightly — Kd values between 2 nM and 50 nM. One activated the receptor.
That peptide was HL4. Its measured Kd was 132 nM, well behind the strongest binders. By the standard logic of an affinity screen — pick the tightest binders, optimize from there — HL4 would have been deprioritized. The team had nothing in their selection method that biased toward functional binders rather than any binders. What rescued HL4 from being discarded was the second step: testing the cellular activity of multiple hits in parallel rather than only the tightest few.
The biological message is that for a complex receptor, tight binding is not a proxy for productive binding. The methodological message is sharper. Affinity-first screens are the industry standard for a reason — they are tractable, fast, and produce numerical rankings. But for hard targets they can throw away exactly the molecules that would have been valuable. The RaPID-ExCells paper is the cleanest evidence in years that on at least some receptors, you have to look at function in parallel with affinity. Function-first selection, the authors argue, deserves to become the default for receptor agonist programs.
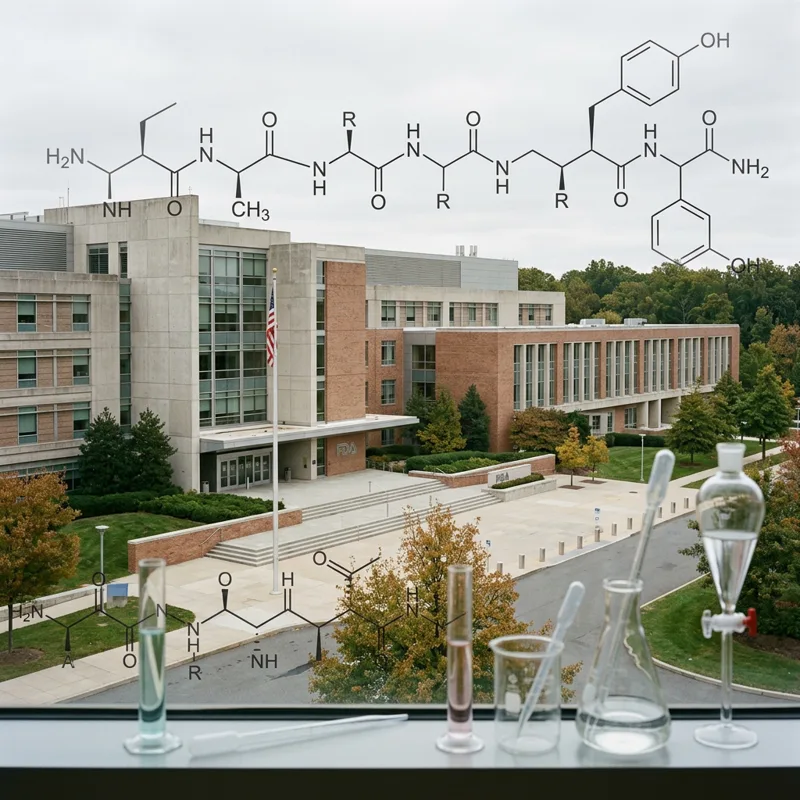
What HL4 Actually Looks Like
HL4 is a cyclic peptide of 19 amino acid residues. Its core sequence is closed through a thioether bond formed during the RaPID translation step, the same chemistry that all RaPID cyclic peptides share. Its activating motif is the five-residue stretch FYLWF at positions 9 to 13: phenylalanine, tyrosine, leucine, tryptophan, phenylalanine. Three aromatic residues flanking a leucine and a tryptophan.
That motif is not an accident. In the 1990s, peptide chemists screening linear libraries against the insulin receptor identified a family of insulin-mimetic linear peptides built around a sequence pattern that came to be written FYXWF, where X is variable. Novo Nordisk's S597 is the best-known example. Those peptides bind a specific allosteric site on the IR ectodomain — site 2 — and produce partial activation. They have never reached the clinic, because their potency was too low and their pharmacokinetics were poor.
HL4 is what you get if you let mRNA display search the cyclic-peptide universe for an activator and the search lands on the same pharmacophore. The aromatic clamp at site 2 is convergent: it shows up in linear chemistry and in cyclic chemistry, in 1990s screens and in 2026 cell-based screens. The implication is that the FYXWF / FYLWF region of insulin-mimetic activity is a real and stable design feature, not a quirk of any one screening method. That gives a chemist with a list of analogs to make a justified place to start.
The team then ran deep mutational scanning with 66 amino acids, of which 47 were non-proteinogenic. Substituting position 9 with para-methyl-phenylalanine or para-fluoro-phenylalanine raised activity by about 50 percent. Substituting position 5 with D-tyrosine was tolerated. The point is not the magnitude of the improvement — 1.5-fold is modest — but the demonstration that the cyclic backbone is permissive enough to accept noncanonical residues, which opens up properties like protease resistance and tunable hydrophobicity that you cannot get from natural amino acids alone.
From Hit to Drug-Shaped Molecule
Insulin itself activates the IR by binding both halves of the dimer in a way that brings them into a productive conformation. A monomeric peptide that binds one half can give partial activation but rarely full agonism. The Suga team made the conceptually obvious next move: link two HL4 molecules together so that one drug molecule can engage both halves.
The construct they describe uses two HL4 monomers connected through PEG11 spacers anchored to a central L-2,3-diaminopropionic acid (L-Dap) residue. The PEG11 spacers give enough length to span the receptor dimer; the L-Dap core gives a defined branching point. The resulting homodimer activates IR autophosphorylation with an EC50 of about 460 nM, compared with about 720 nM for the monomer, and gives roughly 2.5-fold the autophosphorylation signal at 1 µM.
The dimer has two practical properties that matter beyond the EC50 number. Its serum half-life in vitro is around 51 hours, which puts it in the long-acting range for a peptide that has not been formally optimized for pharmacokinetics. And it discriminates between the insulin receptor and the closely related IGF-1 receptor, which is a non-trivial selectivity problem given how similar the two receptors are structurally. The selectivity probably reflects fine geometric matching between the dimer's two HL4 heads and the specific spacing of binding sites on IR versus IGF-1R.
None of this makes HL4 a drug. It is not in any clinical trial. There is no IND. There is no oral formulation. The work shows that the molecule has the kind of properties — selectivity, half-life, defined mechanism — that a chemist would want as a starting point for medicinal optimization. The next steps for the program are unspecified in the paper itself.
The Platform Is the Story
It is tempting to read this paper as "Tokyo and Osaka labs find new insulin-mimetic peptide" and move on. That misses the more important claim. The insulin receptor was chosen as a test case partly because it is hard. If RaPID-ExCells works on IR, the same method should work on a long list of other receptors that have resisted clean peptide drug discovery: many G-protein-coupled receptors with complex conformational landscapes, receptor tyrosine kinases that signal as dimers or higher-order assemblies, transporters and channels that present binding surfaces only in specific membrane environments.
The peptide field has spent the last decade absorbing several discovery-platform shifts at once. Phage display gave way to mRNA display for routine cyclic-peptide selection because mRNA display scales further and tolerates more chemical diversity. Computational design tools, especially those based on AlphaFold and its successors, opened up de novo binder design without screening at all. RaPID-ExCells sits in a third category: a screening method that takes the strengths of mRNA display — library size, chemical diversity, defined chemistry — and pairs them with a biologically realistic readout that computational tools cannot yet reliably replicate.
How widely the platform spreads depends on practical factors. Co-immunoprecipitation works well for some receptors and badly for others. The mild detergent step that preserves IR's dimer state will not preserve every protein's relevant conformation. Some receptors live in lipid rafts or have lipid co-factors that detergent disrupts. The next two years of papers from the Suga lab and from groups that pick up the method will show where RaPID-ExCells lands on the spectrum from "broadly applicable" to "specialty tool for a few targets."
Where This Fits in the Insulin-Mimetic Story
Insulin-mimetic peptides have been a quiet research line since the 1990s. The motivation is straightforward. Insulin works extraordinarily well; the manufacturing, storage, and delivery infrastructure for it does not. Recombinant insulin requires a cold chain. It is injected. Its dosing is unforgiving on either side. A small-molecule or peptide insulin-mimetic that activated IR without those constraints — especially one with oral or extended-release options — would change the practical economics of type 1 and insulin-dependent type 2 diabetes care, particularly outside high-income countries.
Several insulin-mimetic peptide families have been described. S597 and its derivatives bind site 2 on IR. Aptamers that bind IR have been published. Single-chain insulin analogs have been engineered for improved manufacturability. None has reached the clinic as an insulin replacement. Most have run aground on potency, half-life, or manufacturability problems — or on the broader observation that recombinant insulin is, despite its delivery problems, a remarkably good drug and a high bar to clear.
HL4 is unlikely to clear that bar in its current form. Its potency, even as a homodimer, is well below physiological insulin. Its oral availability is unknown. The point of the paper is not that HL4 is the next insulin. The point is that RaPID-ExCells found a functional IR agonist that earlier methods would have missed, with a defined chemistry that supports medicinal optimization, in a single round of selection. If the next IR-targeted peptide that comes out of the same platform has 10-fold better potency and a viable formulation, that is when this paper will be cited as the methodological starting point.
What This Means for Different Audiences
For peptide chemists: RaPID-ExCells is now available as a published method for receptors that have defeated affinity-only screens. The function-first message — test cellular activity on multiple top hits in parallel, not just the strongest binder — applies to almost every peptide drug-discovery program against a complex receptor, not only insulin.
For people in the diabetes and metabolic space: HL4 is not a clinical candidate. It is a proof-of-platform molecule. The realistic interpretation is that there will be a second-generation insulin-mimetic peptide from this lab or a successor lab within a few years that has substantially improved properties. Whether that molecule reaches the clinic depends on potency optimization, formulation work, and the unforgiving comparison against recombinant insulin.
For people watching the broader peptide field: the methods toolkit is widening fast. Computational de novo binder design from groups like the Baker lab and follow-ons. Cell-based mRNA display from the Suga lab. Continued advances in non-natural amino acid incorporation. Most peptide drug programs five years from now will use some combination of these tools. The teams that figure out which method fits which target class will have a significant advantage.
Frequently Asked Questions
Is HL4 a treatment for diabetes?
No, not in its present form. It is an experimental peptide that activates the insulin receptor on cells in a dish. It has not been tested in animals as a glucose-lowering agent in the paper as published, has no IND filing, no clinical trial, and no formulation work for human use. It is a research molecule that demonstrates a new selection method.
How is RaPID-ExCells different from regular RaPID?
Standard RaPID screens cyclic peptide libraries against purified protein targets bound to a surface. RaPID-ExCells screens the same libraries against full-length receptors on live cells, then recovers bound peptides by detergent lysis and co-immunoprecipitation. The difference matters for receptors whose binding surface depends on their membrane environment or quaternary structure, which includes most receptor tyrosine kinases and many GPCRs.
What does "function-first selection" mean in practical terms?
It means picking lead candidates based on what they do on cells rather than how tightly they stick to a purified target. In this paper, the tightest binder was not the activator. The team caught HL4 by testing multiple top hits for cellular activity in parallel, not by trusting the affinity ranking alone. Generalized: assay function early, not after you have already narrowed to the top one or two affinity picks.
Why does the FYLWF motif keep showing up?
The insulin receptor has an allosteric binding site — site 2 — that has a shape complementary to a cluster of aromatic and hydrophobic side chains. When you let a screen search the peptide universe for activators, the chemistry that fits site 2 keeps being rediscovered. Linear-peptide screens in the 1990s found the FYXWF pattern. Cyclic-peptide mRNA display in 2026 finds FYLWF. The motif is essentially the part of an insulin-mimetic peptide where the molecule "grabs" the receptor in the configuration that drives activation. The cyclic backbone gives you better pharmacological properties around that grab.
Is this related to GLP-1 drugs like Ozempic?
Different receptor, different mechanism, different therapeutic area. GLP-1 receptor agonists like semaglutide activate the GLP-1 receptor and produce weight loss and improved glycemic control through that pathway. HL4 activates the insulin receptor directly. They are both peptides, but the targets, the mechanisms, and the clinical use cases are distinct. RaPID-ExCells could in principle be used to find new GLP-1R agonists too — that would be a separate paper.
What is the next milestone to watch?
Two things. First, papers from the Suga lab or labs that license the method applying RaPID-ExCells to other receptors — especially GPCRs and receptor tyrosine kinases that have resisted clean peptide discovery. Second, follow-up papers on the HL4 family — potency optimization, animal pharmacology, oral or long-acting formulation work. Both are reasonable to expect within roughly two years.
Sources
- Kuo Y-H, Mihara E, Takagi J, Suga H. "Direct Selection of Functional De Novo Macrocycles for Activation of On-Cellulo Insulin Receptor." Angewandte Chemie International Edition. Published online March 12, 2026. DOI 10.1002/anie.202526008.
- PubMed listing: PMID 41815006.
- Free full text (PMC): PMC13098473.
- American Peptide Society research feature: "Function-First Macrocycles," May 12, 2026.
- Suga Lab, University of Tokyo: Department of Chemistry, Graduate School of Science.
- Takagi Lab, Institute for Protein Research, Osaka University: IPR Osaka.
- Background on the FYXWF insulin-mimetic motif: Schäffer L et al. "A novel high-affinity peptide antagonist to the insulin receptor." Biochemical and Biophysical Research Communications. 2008. PMID 18585368.
This article reports on preclinical research published in a peer-reviewed journal. HL4 is not a drug. It is not approved for any use in humans. Nothing here is medical advice.
Sources & References
- FDA PCAC Meeting Announcement (July 23-24, 2026)
- PBS: FDA to Weigh Easing Limits on Peptides Favored by RFK Jr.
- BioPharma Dive: FDA Peptides RFK Advisory Committee Restrictions
- RAPS: FDA Considers Adding a Dozen Peptides to Bulk Drug List
- Ars Technica: RFK Jr. Forces FDA to Reconsider 12 Peptides
- ProPublica: Peptide Safety Investigation
- New York Times: Peptide Ban FDA RFK Jr.
- SSRP Institute: FDA Announces Change in Status of 12 Peptides
- CNBC: RFK Jr. Peptides Hims Hers GLP-1
- USA Today: RFK Jr. FDA Peptides Explainer